rdiModel - RDI Models
Description¶
Generate models equating RDI values to true differences in underlying prevalence values
Usage¶
rdiModel(n, ngenes = NULL, baseVects = NULL, nIter = 50, nSample = 20,
units = c("lfc", "pct"), constScale = TRUE)
Arguments¶
- n
- the repertoire size.
- ngenes
- numeric vector indicating the number of genes in each chain.
If
baseVectsis not provided, this parameter will be used to generate a base prevalence vector for each of the genes. - baseVects
- A vector or list of vectors representing the total prevalence of each gene (for each chain) in the dataset. Differential datasets will be created from alterations of this vector. If not provided, a base vector will be randomly . generated at each subsample step containing the number of genes specified by ngenes.
- nIter
- The number of iterations (i.e. number of datasets to generate).
- nSample
- The number of samples to generate for each subsample. Each sample will have a different true fold change, but the same starting vector
- units
- String; either “lfc” or “pct”, depending on what transform was used in the original RDI calculations. See Details.
- constScale
- logical; if
TRUE, vdjCounts will be scaled such that the sum of each column will be equal to 500 (an arbitrary constant). Otherwise, the columns will be scaled to the average count of all the columns.
Value¶
A list containing three objects:
| fit |
an object of class "smooth.spline", based on a spline
model with the true difference (lfc or pct) as the independent (x)
and RDI as the dependent (y). Used for converting from true difference
to RDI. |
| rev.fit |
an object of class "smooth.spline".
The opposite of fit. Used for converting from RDI to true difference.
|
Details¶
This method uses simulated sequencing datasets to estimate the RDI values for datasets with a known true deviation.
Briefly, a baseline probability vector (either randomly generated or supplied by the
baseVects parameter) is randomly perturbed, and the difference between the
baseline vector and the perturbed vector is calculated. Then, nSample sequencing
datasets of size n are randomly drawn from both the baseline vector and the perturbed
vector, and the RDI distance between all datasets calculated. This process is repeated
nIter times, resulting in a dataset of RDI values and matched true differences.
A set of spline models is then fit to the data: one from RDI to true difference, and
another from true difference to RDI value, allowing for bi-directional conversions.
If a baseline probability vector is not provided, one will be generated from an empirical model of gene segment prevalence. However, for best performance, this is not recommended. Estimates of true fold change is very sensitive to the distribution of features in your count dataset, and it is important that your baseline vector match your overall dataset as accurately as possible. The best baseline vector is almost always the average feature prevalence across all repertoires in a dataset, although manually generated baseline vectors may also work well.
The units used for the RDI model should always match the units used to generate your RDI values. For more details on units, refer to the details of calcRDI.
Examples¶
#create genes
genes = sample(letters, 10000, replace=TRUE)
#create sequence annotations
seqAnnot = data.frame(donor = sample(1:4, 10000, replace=TRUE))
#calculate RDI
d = rdi(genes, seqAnnot)
##create a "baseVect" with the same probability as our features
##since we sampled uniformly, the base vector has equal probability
baseVect = rep(1/length(letters),length(letters))
##generate an RDI model
m = rdiModel(attr(d, "nseq"), baseVects=baseVect)
##plot the spline model
plot(m$fit, xlab="log fold change",ylab="RDI",type='l')
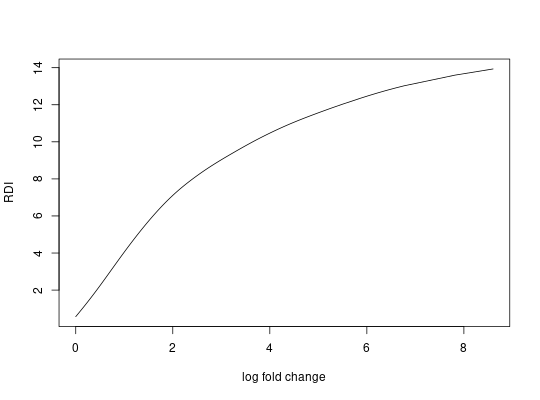
##convert RDI to log fold change
mean = predict(m$rev.fit, d)$y
mean[mean<0] = 0