calcRDI - Calculate repertoire distances
Description¶
Calculate repertoire distances from a matrix of vdjCounts
Usage¶
calcRDI(vdjCounts, distMethod = c("euclidean", "cor"), subsample = TRUE,
nIter = 100, constScale = TRUE, units = c("lfc", "pct"), ...)
Arguments¶
- vdjCounts
- a matrix of repertoire counts, as created by calcVDJCounts
- distMethod
- one of c(“euclidean”,”cor”) determining how to calculate the distance from the matrix of vdj counts. See Details.
- subsample
- logical; if true, all repertoires are subsampled to be equal size.
- nIter
- value defining how many times the subsampling should be repeated. Only used if subsample is TRUE.
- constScale
- logical; if
TRUE, vdjCounts will be scaled such that the sum of each column will be equal to 500 counts (an arbitrary constant). Otherwise, the columns will be scaled to the average count of all the columns. - units
- One of “lfc” or “pct”. This determines the method used for transforming the repertoire counts. See Details.
- …
- additional parameters; these are ignored by this function.
Value¶
A dissimilarity structure containing distances between repertoires, averaged across each subsampe run. In addition to the standard attributes in a dist object, three additional attributes are defined as follows:
| ngenes |
| integers, the number of genes in each column of “genes” that were included in at least one repertoire. |
| nseq |
| integer, the number of sequences used after subsampling the repertoires. If subsample=FALSE, this is not
defined. |
Details¶
There are two options for distance methods, “euclidean” and “cor”. Euclidean refers to
standard euclidean distance, and is the standard for the RDI measure described in
(Bolen et al. Bioinformatics 2016). In contrast, cor refers to a correlation-based
distance metric, where the distance is defined as (1-correlation) between each
column of vdjCounts.
The units parameter is used to determine the transformation function for the
repertoire counts. If units='lfc' (default), then the arcsinh transformation
is applied to the count matrix, resulting in a distance metric which
will scale with the average log fold change of each gene. In contrast,
units='pct' will result in no transformation of the count matrix, and distances
will be proportional to the average percent change of each gene, instead. Note that
“units” is a bit of a misnomer, as the distance metric doesn’t actually represent the
true log-fold or percent change in the repertoires. In order to actually estimate
these parameters, refer to the rdiModel and convertRDI
functions.
Examples¶
#create genes
genes = sample(letters, 10000, replace=TRUE)
#create sequence annotations
seqAnnot = data.frame(donor = sample(1:4, 10000, replace=TRUE),
cellType = sample(c("B","T"), 10000, replace=TRUE)
)
##generate repertoire counts
cts = calcVDJcounts(genes,seqAnnot)
##calculate RDI
d = calcRDI(cts)
##calculate RDI in percent space
d_pct = calcRDI(cts,units="pct")
##convert RDI to actual 'lfc' estimates and compare
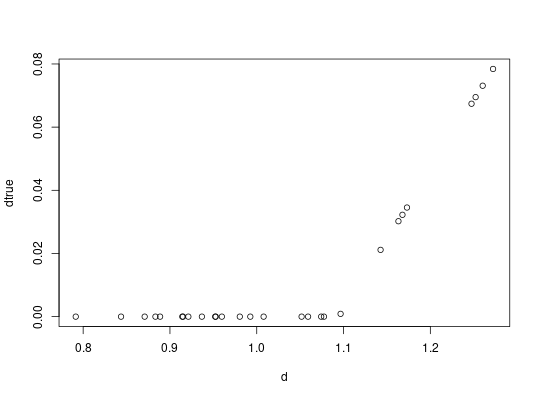
dtrue = convertRDI(d)$pred
plot(d, dtrue)